Close
Skrining podrazumeva sistematsku primenu medicinskih testova u cilju ranog otkrivanja bolesti, u trenutku kada simptomi bolesti nisu prisutni. Radi se kod osoba koje nisu tražile lekarsku pomoć, radi otkrivanja pojedinaca s rizikom od određenog poremećaja koji je dovoljno veliki da opravdava dalje ispitivanje i preventivno delovanje. Pošto se ispitivanje sprovodi pre nego što osoba ispolji simptome, lečenje može da započne pre nego što se razvije klinička slika i pre nego što nastanu komplikacije.
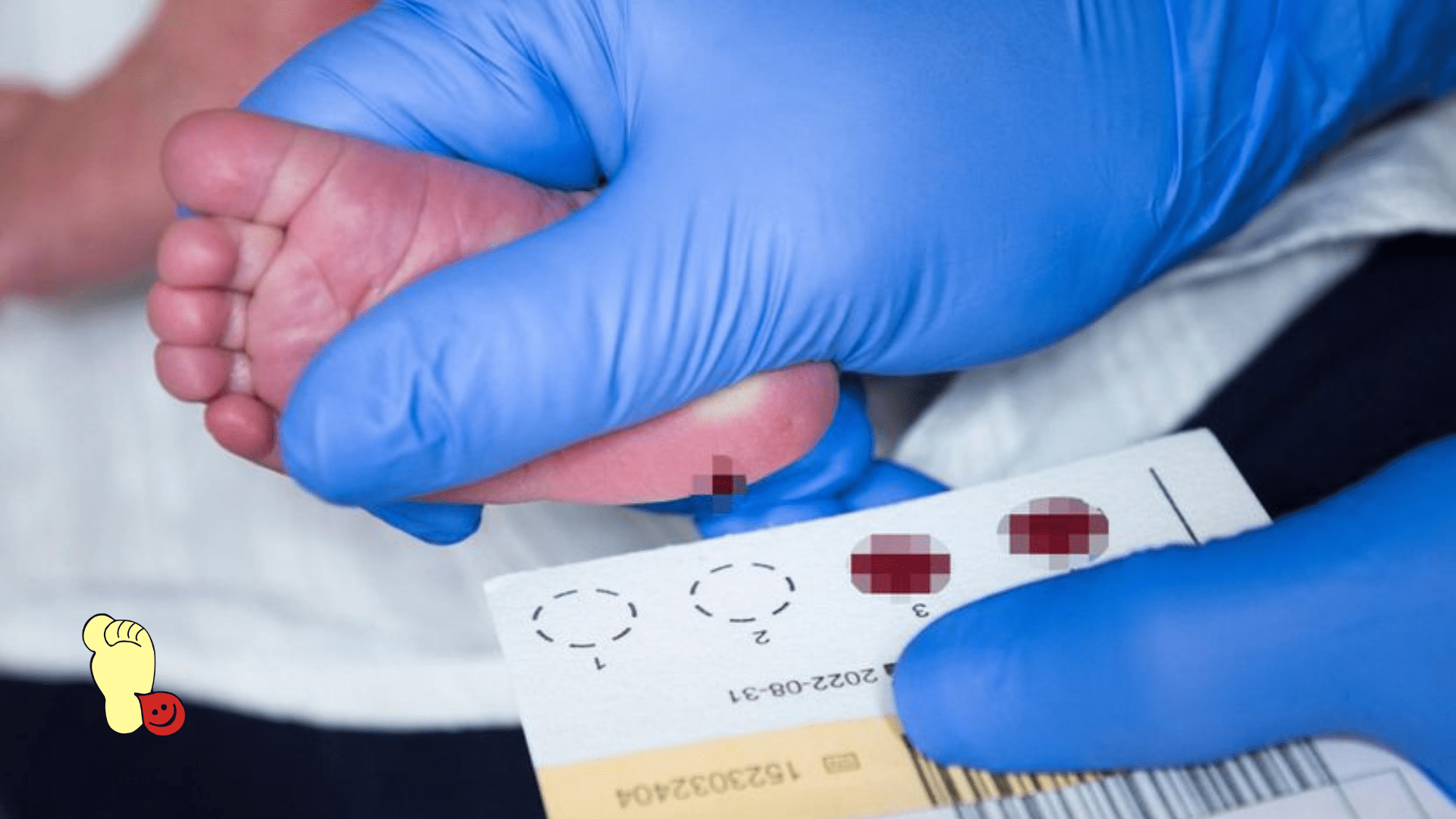
Sva deca rođena u Republici Srbiji prolaze neonatalni skrinig. Krv za skrining se uzima u porodilištu, a analize se rade u laboratorijama van porodilišta. U Centralnoj Srbiji uzorci se šalju u laboratoriju Instituta za zdravstvenu zaštitu majke i deteta Srbije „Dr Vukan Čupić“, a u Vojvodini u laboratoriju Instituta za zdravstvenu zaštitu dece i omladine Vojvodine. Za decu iz cele Srbije, obrada uzoraka za skrining na spinalnu mišićnu atrofiju se vrši na jednom mestu, tj. na Biološkom fakultetu u Beogradu.
Postoji veliki broj bolesti i stanja za koja se može sprovoditi neonatalni skrining. Broj bolesti na koje se sprovodi skrining razlikuje se od zemlje do zemlje.
Ako rezultat neonatalnog skrininga nije normalan, to još uvek ne znači da dete ima bolest za koju se skrining vrši. U tom slučaju, neophodno je uraditi dodatne testove, kako bi se utvrdilo da li dete ima bolest za koju se skrining sprovodi. U većini slučajeva, deca koja imaju bolest po rođenju izgledaju potpuno normalno. Neonatalni skrining pomaže da se bolest otkrije pravovremeno, tako da terapija može da se započne što pre i na taj način spreče komplikacije.
U Srbiji se sprovodi neonatalni skrining analizom kapi krvi koja se uzima iz pete novorođenčeta u porodilištu na 4 bolesti: fenilketonuriju, kongenitalni hipotiroidizam, cističnu fibrozu i spinalnu mišićnu atrofiju. U toku 2025. godine, iz iste kapi, treba da započne skrining i na kongenitalnu adrenalnu hiperplaziju. Takođe, u brojnim, ali ne svim porodilištima, novorođenoj deci se radi i skrining sluha.
Za bliže informacije o oboljenjima, kliknite na linkove ispod.
Fenilketonurija je bolest u kojoj aminokiselina fenilalnin (sastavni deo svih proteina) ne može da se razgradi, što dovodi do oštećenja nervnog sistema, ozbiljne mentalne zaostalosti i epilepsije.
Uzrok je genetički. Zdravi roditelji nose sklonost za oboljevanje, a verovatnoća u svakoj trudnoći da dete oboli je 25%. Ako se desi, genetički poremećaj dovodi do smanjene aktivnosti enzima fenilalanin hidroksilaze koji je neophodan za razlaganje fenilalanina.
Novorođena deca sa fenilketonurijom nemaju simptome bolesti i ne razlikuju se od ostale zdrave novorođenčadi. Ako bolest ne počne da se leči u prvom mesecu života, prvi simptomi se javljaju tek posle nekoliko meseci života u smislu neurološkog propadanja.
Fenilketonurija se otkriva neposredno po rođenju, u okviru neonatalnog skrining programa, u Srbiji i gotovo u svim zemljama sveta. Organizovano otkrivanje bolesti u našoj zemlji traje više od četrdeset godina. Uzorak krvi novorođenog deteta se u trećem danu života (najranije 48 sati od rođenja, isključivo u porodilištu), kada smo sigurni da je dete jelo jer je to jedini način da se fenilalanin unese u organizam. Uzorci krvi na filter papiru se šalju u centralnu laboratoriju gde se određuje koncentracija fenilalnina. Rezultati analiza se čuvaju u arhivi centralne laboratorije, kao i u porodilištu, na način koji garantuje poštovanje principa lekarske tajne.
U proseku se u Srbiji otkrije dva do tri deteta godišnje sa fenilketonurijom.
U slučaju da su rezultati ispitivanja normalni, roditelji se o tome ne obaveštavaju.
Ako rezultat pokaže više koncentracije fenilalnina to još uvek ne označava bolest, ali se ta deca pozivaju u Institut radi provere i dopunskih analiza.
Da, i to veoma uspešno. Naime, simptomi fenilketonurije se ne javljaju ako se terapija počne na vreme i dobro sprovodi.
Fenilketonurija se leči dijetom. Dijeta podrazumeva ograničeni unos proteina uz
korišćenje mešavina aminokiselina bez fenilalnina. Dijeta mora biti kontrolisana i
vođena od strane stručnih lica koja se bave fenilketonurijom. Roditelji moraju da se dobro
obuče u sprovođenju dijete.
Kontrole se uglavnom obavljaju slanjem uzoraka krvi na filter papiru (merenjem
koncentracije fenilalnina), a ređe dolaskom u Institut na ambulantni pregled.
Kao svaka druga genetička bolest fenilketonurija traje ceo život, te se preporučuje doživotna dijeta koja s vremenom postaje manje zahtevna.
Za sada se fenilketonurija ne otkriva pre rođenja, s obzirom da nakon postavljene rane
dijagnoze mogu da se spreče simptomi posebno organizovanom ishranom.
Striktno pridržavanje dijete omogućava normalan rast i razvoj, a s vremenom dijeta postaje
sastavni deo života svake osobe sa fenilketonurijom. Fenilketonurija nije prepreka
normalnom životu.
Kongenitalni hipotiroidizam je stanje u kome u organizmu nema dovoljno hormona štitaste žlezde. Beba se rađa sa ovim stanjem (reč „kongenitalno“ znači urođeno, tj. da je osoba rođena sa tim).
Štitasta (tiroidna) žlezda nalazi se u predelu vrata i ona luči tiroidne hormone. Ovi hormoni važni su za pravilan rast i razvoj, održavanje telesne temperature, kao i za brojne metaboličke procese. Ukoliko štitasta žlezda ne luči dovoljno hormona govorimo o „hipotiroidizmu“.
Uzroci mogu biti razni – štitasta žlezda može da nedostaje, može biti manja nego uobičajeno ili formirana, ali na drugom mestu od uobičajenog. Ponekad, problem može da bude prouzrokovan i promenama na genetičkom nivou. Tada poremećaj može da se vidi kod više članova jedne porodici.
Pored toga, smanjena količina hormona štitaste žlezde može se naći kod novorođenčadi čije majke ne koriste dovoljno joda u ishrani (što je u Srbiji izuzetno retko) ili kod onih čije majke uzimaju prekomerne količine joda ili određene lekove. U tim slučajevima obično dođe do postepenog oporavka.
Najveći broj novorođenčadi nema nikakve simptome i uobičajeno se ponašaju. Većina novorođenčadi u prve 2-3 nedelje ima dovoljno hormona štitaste žlezde koji su kroz posteljicu prešli iz krvi majke u krv deteta.
Ako simptomi postoje, to može biti nešto od sledećeg: žutica, manja aktivnost (beba neuobičajeno dugo spava), usporeni ili retki pokreti, promukao plač, problemi sa hranjenjem, konstipacija (zatvor), neuobičajen izgled – uvećan jezik, napetost oko pupka, široko otvorena velika fontanela, otvorena mala fontanela, smanjen mišićni tonus (hipotonija), suva koža, hladni periferni delovi ruku ili nogu.
U Srbiji (kao i većini zemalja u svetu) se od 1983. godine sprovodi neonatalni skrining na kongenitalni hipotiroidizam. On podrazumeva uzimanje jedne kapi krvi iz pete novorođenčeta i određivanje vrednosti hormona iz tog uzorka. Uzorak krvi se uzima najranije u 48. satu života, isključivo u porodilištu. Rezultati analiza se čuvaju u arhivi centralne laboratorije, kao i u porodilištu, na način koji garantuje poštovanje principa lekarske tajne.
Ako se rano (odmah po dijagnostikovanju) započne sa davanjem terapije, sprečavaju se moguće posledice i dete se u potpunosti normalno razvija. Uz terapiju, deca i odrasli sa kongenitalnim hipotiroidizmom imaju sasvim normalan život.
Veoma je važno da se održavaju normalni nivoi hormona štitaste žlezde u svim uzrastima, ali je to naročito važno kod odojčadi i male dece zato što su hormoni štitaste žlezde vrlo bitni za rast i razvoj deteta, kao i za funkcionisanje i razvoj mozga, te je neophodno da se roditelji striktno pridržavaju terapije koju je propisao endokrinolog.
Kongenitalna adrenalna hiperplazija (KAH) je urođeni poremećaj nadbubrežnih žlezda koji dovodi do smanjene produkcije određenih hormona i prekomerne produkcije drugih. Beba se sa ovim poremećajem rađa, a KAH najčešće nastaje zbog urođenog nedostatka enzima koji je neophodan za normalnu sintezu hormona u nadbubrežnim žlezdama.
Nadbubrežne žlezde se nalaze iznad oba bubrega i luče nekoliko veoma važnih hormona kao što su kortizol, aldosteron i androgeni (muški polni hormoni). Kod KAH-a, najčešće nedostaje enzim 21-hidroksilaza, što dovodi do nedostatka kortizola i/ili aldosterona i velikog viška muških polnih hormona.
Postoji više oblika KAH-a, ali najteži je takozvani „oblik sa gubitkom soli“. Simptomi se mogu javiti u prvim danima života i uključuju: povraćanje, loše napredovanje, pospanost, odbijanje obroka, dehidraciju, loše opšte stanje.
Kod devojčica je već na rođenju uočljiv drugačiji izgled spoljašnjih genitalija, koje često liče na muške zbog velikog viška muških polnih hormona. Kod dečaka spoljašnje genitalije su tipične muške, pa se bolest otkriva kasnije, ali uglavnom u prve 2 nedelje života.
Ne može se sprečiti, jer je genetski poremećaj, ali se mogu sprečiti komplikacije ranim otkrivanjem i pravilnim lečenjem.
Cistična fibroza je nasledna bolest koja zahvata više organskih sistema u telu, ali najviše pluća, pankreas i jetru. Bolest nastaje usled poremećaja rada jonskog kanala koji je zadužen za prolazak hlora kroz membranu ćelija.
Najveći broj novorođenčadi nema posebne simptome i deluju potpuno zdravo.
Ponekad se dešava da gust crevni sadržaj blokira normalan prolazak stolice kroz creva izazivajući njihovu opstrukciju i nadimanje stomaka, stanje koje se naziva mekonijalni ileus. Ono zahteva hirurško lečenje već u prvim danima života i jasno pobuđuje sumnju kod lekara da novorođenče boluje od cistične fibroze.
Bebe obolele od cistične fibroze mogu imati produženu neonatalnu žuticu, češće respiratorne infekcije ili slabije napredovanje u telesnoj masi, uz pojavu masnijih stolica.
Promene na plućima, pankreasu i jetri su počele da se razvijaju kod bebe dok je još bila u majčinom stomaku, stoga je od velike važnosti postaviti dijagnozu cistične fibroze i pre nego što se jave specifični respiratorni ili crevni simptomi, da bi se odgovarajuća terapija započela što ranije. Na vreme primenjena terapija usporava prirodni tok bolesti i sprečava rani nastanak promena na pomenutim organima bebe.
U našoj ustanovi se od aprila 2022. godine sprovodi neonatalni skrining na cističnu fibrozu, iz uzorka kapilarne krvi na filter papiru uzetog iz pete novorođenčeta dok je boravilo u porodilištu. Ovaj uzorak se iz svih porodilišta u Srbiji šalje u Institut na dalju analizu. Izuzetak su porodilišta iz autonomne pokrajine Vojvodina, koja samostalno sprovodi neonatalni skrining na svojoj teritoriji.
U slučaju da je ovaj prvi nalaz pozitivan, o tome obaveštavamo porodilište iz kog je prvi uzorak i poslat. Nakon kontaktiranja porodice savetuje im se dolazak u Institut radi ponavljanja analize iz novog uzorka kapilarne krvi na filter papiru, najkasnije do 21-og dana života novorođenčeta. Ukoliko i ovaj drugi nalaz bude pozitivan, to znači da postoji verovatnoća da beba zaista i boluje od cistične fibroze.
Za postavljanje definitivne dijagnoze potreban je dopunski znojni test, kojim se analizira koncentracija hlorida u znoju bebe. Test se izvodi kasnije, od trećeg meseca života, o čemu roditelji budu na vreme obavešteni putem telegrama. U njemu se jasno navodi da je razlog njihovog pozivanja sumnja da postoji verovatnoća da njihovo dete boluje od cističe fibroze, kao i datum zakazanog testiranja i ime lekara koji će ih tog dana primiti.
Ako rezultat znojnog testa potvrdi sumnju da je beba obolela od cistične fibroze rade se i dodatne molekularne genetske analize iz uzorka venske krvi da bi se utvrdile tačne mutacije gena kod bebe. Ova dopunska genetska analiza je neophodna radi daljeg planiranja lečenja ovih pacijenata.
Spinalna mišićna atrofija (SMA) je urođeno oboljenje mišića i nerava. Kod ovog oboljenja dolazi do ranog propadanja nervnih ćelija koje inervišu mišiće, a zovu se motorni neuroni. One se nalaze u kičmenoj moždini i u mozgu. Zbog rane smrti motornog neurona dolazi do propadanja mišića i njihove atrofije, odnosno gubitka mišićne mase i snage. Tokom vremena bolest napreduje, zbog čega SMA spada u grupu teških, progresivnih neuromišićnih bolesti. To je nasledna bolest i potrebno je da razumemo njenu genetičku osnovu, kako bi nam bila jasnija priroda oboljenja i različitost u ispoljavanju.
Spinalna mišićna atrofija je prouzrokovana nedovoljnom funkcijom proteina neophodnog za preživljavanje motornog neurona, čija je skraćenica SMN protein od engleskog naziva “survival motoneuron protein“. Ovaj protein nastaje prepisivanjem gena koji ima istu oznaku, SMN gen i nalazi se na 5. hromozomu. Kod zdravih osoba postoje dva SMN gena, koji se označavaju kao SMN1 i SMN2 gen. Po pravilu, postoje dve kopije SMN1 gena i više (2 do 6) kopija SMN2 gena. Prepisivanjem SMN1 gena nastaje 100% funkcionalan SMN protein, značajan za život motornih nervnih ćelija. Prepisivanjem drugog, tzv. SMN2 gena nastaje protein koji je slabo funkcionalan. Samo 10-15% ovog proteina ima aktivnu funkciju u životu nervnih ćelija. Ljudi koji boluju od SMA imaju „grešku“ u obe kopije SMN1 gena, jedne nasleđene od oca i druge, nasleđene od majke. Ta greška je kod 97-98% obolele dece u vidu nedostatka nekog segmenta SMN1 gena, stručno rečeno delecije u samom genu. Mnogo ređe (kod 2-3% obolelih) reč je o maloj, tačkastoj mutaciji, koja može „promaći“ klasičnim genetičkim testovima i dijagnostikuje se tek sa pojavom simptoma bolesti drugačijim tehnikama (metodom sekvenciranja gena). S obzirom da SMN2 gen ima „slabu“ funkciju, on nikako ne može da zameni SMN1 gen i potpuno nadomesti njegovu ulogu, iako oboleli mogu imati veći broj kopija ovog SMN2 gena. Međutim, značaj SMN2 gena je u određivanju težine bolesti, što zavisi od broja njegovih kopija.
Ukoliko osoba ima jedan normalan SMN1 gen, a drugi sa greškom, tj. nedostatkom jednog dela, ona neće imati bolest. Takve ljude nazivamo zdravim prenosiocima bolesti. Ako oba roditelja nose gen sa greškom, rizik da se rodi dete obolelo od SMA iznosi 25%. Mogućnost da dobiju dete koje ima samo jednu kopiju SMN1 gena i prenosilac je bolesti kao i sami roditelji iznosi 50%. Učestalost ljudi u opštoj populaciji koji su zdravi prenosioci bolesti nije mala i iznosi 1 na 40-60 osoba.
SMA tip 0: Deca na rođenju uglavnom nemaju znake bolesti. Retko deca sa SMA mogu imati početak bolesti pre rođenja i da već na rođenju ispolje mlitavost (hipotoniju), mišićnu slabost, oslabljene spontane pokrete ekstremiteta. Oni vrlo brzo razvijaju poremećaj disajne funkcije, do nemogućnosti samostalnog disanja i žive kratko. Za ovu decu kažemo da imaju „nulti“ tip SMA, koji se izdvaja kao peti oblik ove bolesti i javlja se kod oko 1% obolelih. 2
SMA tip 1 (Verdnig-Hofmanova bolest): Najveći broj obolelih, oko 60%, ima SMA tip 1 ili Verdnig-Hofmanovu bolest, koja je dobila ime po autorima koji su je među prvima opisali. Ova deca se rađaju bez simptoma i znakova bolesti. Prvi znaci bolesti se javljaju u prvih 6 meseci života, a najčešće u 2.-3. mesecu. Slabost kod nelečene dece vrlo brzo napreduje, najpre se smanjuje spontana pokretljivost nogu, a potom ruku i mišića trupa, važnih za disanje, tako da deca ne mogu samostalno da dišu i potrebna im je potpora mašinom za disanje. Imaju teškoće sa gutanjem hrane. Nikada ne steknu sposobnost samostalnog sedenja. Dok nije bilo lekova, deca sa SMA tip 1 su umirala pre 2. rođendana. Oni imaju po 1 do 2 kopije SMN2 gena.
SMA tip 2 ili Duboviceva bolest: Ako gubitak stečenih motoričkih funkcija kod deteta usled slabosti mišića nastupi od 7. do 18. meseca života, govorimo o tipu 2 SMA. Ova deca nauče da sede, ali nikada ne prohodaju. Kod najtežih formi SMA tip 2, deca tokom vremena gube funkciju sedenja. Poseban problem za njih predstavljaju deformiteti kičmenog stuba. Njihov životni vek je duži od 2 godine, a oko 70% živi do 25. godine života. Oni uglavnom imaju po 3 kopije SMN2 gena.
SMA tip 3 ili Kugelberg–Velanderova bolest: Deca koja imaju 3–4 kopije SMN2 gena prohodaju, ali hod postaje nestabilan usled razvoja mišićnih slabosti u periodu posle 18. meseca života. Ako se simptomi ispolje od 18. do 36. meseca, govorimo o tipu 3A bolesti, a ukoliko nastanu posle 36. meseca života, u pitanju je 3B podtip bolesti. Bolesnici sa SMA tip 3 imaju normalan životni vek.
SMA tip 4 ili adultni oblik SMA: U pitanju su odrasli pacijenti, kod kojih tegobe u vidu slabosti donjih ekstremiteta, teškoća sa stajanjem i hodom nastaju od 20. do 30. godine. Funkciju samostalnog hoda uglavnom sačuvaju. Prosečni životni vek ovih bolesnika se ne razlikuje od opšte populacije i oni poseduju po 4 ili više kopija SMN2 gena.
U Srbiji, kao i u većini zemalja u svetu, do 2023. godine bolest se dijagnostikovala na osnovu kliničkog pregleda deteta. Kod sumnje na SMA deca su testirana genetičkim testovima, radi potvrde kliničke dijagnoze. Ovi testovi su se radili u dve referentne laboratorije u Srbiji, u genetičkoj laboratoriji Instituta za zdravstvenu zaštitu majke i deteta Srbije i na Biološkom fakultetu Univerziteta u Beogradu, kao i danas. Od 15. septembra 2023. godine sprovodi se skrining novorođenčadi u svim porodilištima u Srbiji na SMA, uzimanjem uzorka krvi na filter papiru koji se šalje u Biološki fakultet Univerziteta u Beogradu. U genetičkoj laboratoriji Biološkog fakulteta se radi genetičko testiranje i ovo je jedina bolest kod nas gde je neonatalni skrining zapravo genetički test. Rezultati testiranja se čuvaju u arhivi centralne laboratorije, kao i u porodilištu, na način koji garantuje poštovanje principa lekarske tajne.
Da, moguće je dobiti lažno negativan nalaz. To se dešava u situacijama kada osoba poseduje samo jednu umesto dve kopije SMN1 gena, i u toj jednoj kopiji poseduje tačkastu mutaciju, koja „ruši“ normalnu funkciju ovog gena, odnosno proteina dobijenog njegovim prepisivanjem. Ta mogućnost iznosi 2–3%. U ovim situacijama bolest se otkriva sa pojavom prvih simptoma.
U slučaju da su rezultati testiranja negativni, odnosno normalni, roditelji se ne obaveštavaju o tome.
Ako rezultat ukazuje da postoji SMA kod bebe, analiza se još jednom ponavlja, kako bi se sa sigurnošću potvrdilo oboljenje. Određuje se i broj kopija SMN2 gena, što je značajno za težinu oboljenja, odnosno ukazuje na to koji će se tip bolesti razviti kod deteta. Roditelji se pozivaju na razgovor i dijagnozu im saopštava lekar, neuropedijatar ili dečiji neurolog koji se bavi lečenjem bolesnika sa SMA.
Spinalna mišićna atrofija se javlja kod jednog od 6 000 do 10 000 živorođene dece. Zahvaljujući neonatalnom skriningu, u Srbiji je procenjeno da jedno od 9066 novorođene dece boluje od SMA. Dakle, u proseku se na godišnjem nivou rodi 5–6 beba sa ovom bolešću.
Postoje lekovi za lečenje SMA, koji još uvek nisu terapija potpunog izlečenja, već menjaju
prirodni tok bolesti. Zahvaljujući novim, inovativnim lekovima i najteži bolesnici sa
SMA tip 1 dostižu pojedine faze motoričkog razvoja, kao što su sedenje, stajanje i hod, što je
ranijih decenija bilo nezamislivo. Započinjanjem terapije veoma rano, pre prvih simptoma
bolesti, daje se obolelom detetu mogućnost boljeg života, sa manje poteškoća i
komplikacija. Svaki dan kašnjenja u terapiji znači umiranje novih motornih neurona i
razvoj slabosti mišića.
Za decu sa SMA je najvažnije da se lečenje započne što ranije, pre nego li se pojave prvi znaci mišićne slabosti. Često čujemo krilaticu, koja je tačna, da je „motorni neuron = vreme“, koja odražava važnost ranog započinjanja terapije.
Prvi lek koji je odobren za terapiju SMA je nusinersen. Njegova primena u Americi je započeta 2016. godine, u Evropi 2017. godine, a u Srbiji jula 2018. godine. To je mali molekul (tzv. oligonukleotid), koji se aplikuje u kičmeni kanal i direktno deluje na motorne neurone prednjih rogova kičmene moždine, sprečavajući njihovo izumiranje. On poboljšava funkciju SMN2 gena, delujući da se sintetiše mnogo više funkcionalnog SMN2 proteina, u cilju da se nadomesti nepostojeći SMN1 protein.
Drugi lek slično deluje kao i nusinersen, zove se risdiplam i primenjuje se u vidu sirupa.
Treća dostupna terapija je genska terapija („Zolgensma“), koja se primenjuje u vidu jednokratne infuzije.
Ne smemo zaboraviti značaj fizikalne terapije i multidisciplinarni pristup u lečenju dece sa SMA, bez čega primena medikamentne terapije neće dati zadovoljavajuće rezultate.
U najranijem uzrastu neurološke kontrole su česte, a zavise od kliničkog oblika bolesti i opšteg stanja deteta. Veoma je važno da se prate faze motoričkog razvoja deteta. Oboleli od SMA imaju normalnu inteligenciju, mogu biti i natprosečno inteligentni.
Spinalna mišićna atrofija je genetičko oboljenje, zapisano u genetskom kodu osobe koja ga nosi i samim tim je bolest doživotna.
Spinalna mišićna atrofija može da se prevenira u porodici koja ima obolelo dete, tako što će se uraditi prenatalno genetičko testiranje u prvim mesecima trudnoće majke. Trudnica treba da se na vreme obrati genetičkom savetovalištu.
Dete sa SMA može imati daleko bolju prognozu za razvoj i život, zahvaljujući primeni novih, inovativnih lekova, koji modifikuju prirodni tok bolesti i prevode teži u lakši oblik bolesti.
Pojam oštećenja sluha obuhvata nagluvost i gluvoću. Nagluvost podrazumeva ograničenje slušne sposobnosti, a gluvoća odsustvo slušne funkcije. Oštećenja sluha obuhvataju širok spektar, ali najvažnija su trajna, kongenitalna, tj. urođena ili rano nastala, obostrana teškog stepena. Trajno oštećenje sluha kod dece podrazumeva obostrano senzorineuralno oštećenje sluha sa pragom sluha ispod 40 dB (srednja vrednost za čisti ton na karakterističnim frekvencijama). Oštećenja sluha mogu biti jednostrana i obostrana, mogu biti prolazna i trajna, nasledna i stečena a u odnosu na prirodu progresivna i neprogresivna. Zavisno od vremena nastanka odnosno ispoljavanja mogu biti prenatalna, perinatalna i postnatalna. U zavisnosti od vremena ispoljavanja u odnosu na razvoj govorne funkcije, oštećenja sluha dele se na prelingvalna, nastala pre razvoja govora i postlingvalna, nastala polsle uspostavljanja govorne funkcije.
Senzorineuralno oštećenje sluha (SNOS) je posledica oštećenja finog senzornog tkiva unutrašnjeg uva ili čitavog slušnog nerva. Brojni su faktori rizika za nastanak oštećenja sluha. Prihvaćena je sledeća klasifikacija:
Oštećenje sluha se u većini slučajeva ne vidi. Novorođenče ne pokazuje nikakve kliničke znakove, ima uobičajeno ponašanje. Tek se vremenom može primetiti slabija reakcija na zvuk.
Kod malog broja dece se može reći da je vidljivo, zapravo onda kada odsustvo ili značajno umanjenje ušne školjke ukazuju na postojanje oštećenja sluha.
Urođeno oštećenje sluha se otkriva neonatalnim skriningom. Skrining se sprovodi u porodilištima snimanjem tranzitornih otoakustičkih emisija (TEOAE), koje ukazuju na postojanje oštećenja sluha u spoljašnjem, srednjem i unutrašnjem uvu. Ono što one ne otkrivaju, jeste oštećenje slušnog živca i i drugih slušnih puteva koji vode informaciju o zvuku do kore velikog mozga. Ovi, poslednji navedeni poremećaji se javljaju izuzetno retko. Merenje OAE je objektivno, neinvazivno, brzo, ponovljivo i precizno. U osnovi OAE daju objektivan uvid u funkcionisanje celog presinaptičkog auditivnog sistema. Na sreću, najveći broj slušnih oštećenja posledica je patoloških promena upravo u ovom delu auditornog aparata. Tranzitorne OAE, sa širokopojasno frekventnim, kratkotrajnim zvučnim stimulusom koji aktivira celu kohleu jer u sebi sadrži sve frekvencije, u najširoj su upotrebi. Veoma su osetljiva metoda i ne mogu se registrovati sa onih područja kohlee gde je prag sluha veći od oko 30 dB. Otoakustičke emisije se u kliničkoj praksi upotrebljavaju više od 30 godina. Ovom metodom se najbolje može ispitati funkcija senzornih slušnih ćelija i diferencirati njihovo oštećenje od oštećenja proksimalnih nervnih auditornih struktura i funkcija.
Za novorođenčad i malu odojčad, do šest meseci, fiziološka merenja su pristup izbora. Čak i čujuće, zdrave bebe ne obezbeđuju realne bihejvioralne odgovore na zvuk pre navršenih šest meseci starosti. Starija deca, međutim, mogu biti testirana efikasno i efektivno i fiziološkim i bihejvioralnim merenjima.
Slušno oštećenje ne podleže obavezi prijavljivanja u Srbiji i to dovodi do nedostatka podataka o pravom broju , ali procena je da se godišnje u Srbiji rađa oko 150 dece sa trajnim oštećenjem sluha. Pre svega se misli na trajno senzorineuralno oštećenje sluha (TSNOS). Njegovo otkrivanje i jeste cilj neonatalnog skrininga. Oštećenje sluha predstavlja najčešće senzorno oštećenje u ljudi, sa značajnim socijalnim i psihološkim implikacijama. Procenjeno je da približno 20% ljudske populacije starije od 18 godina pati od neke forme slušnog oštećenja.
Na nivou opšte populacije, troje od 1000 novorođene dece ima oštećenje sluha, a od njih jedno ima duboko kongenitalno trajno senzorineuralno oštećenje sluha, a u populaciji dece koja su boravila u odeljenjima za intenzivnu negu ta učestalost iznosi 1:50.
Normalan rezultat skrininga koji se sprovodi samo snimanjem otoakustičkih emisija ukazuje na to da je uredna funkcija spoljašnjeg, srednjeg i unutrašnjeg uva. O funkciji slušnog nerva ne daje informaciju. To znači da potvrda uredne funkcije sluha nije kompletna. Ne otkriva se auditivna neuropatija, spektar poremećaja, poseban senzorineuralni entitet, koji se javlja znatno češće nego što se ranije smatralo, čak u oko 10 % pacijenata sa trajnim senzorineuralnim oštećenjem sluha. Hiperbilirubinemija (žutica) i hipoksija (manjak kiseonika) u novorođenačkom uzrastu su najvažniji riziko faktori za njen nastanak. Kod neke dece je to nasledni poremećaj, a javlja se u sklopu polineuropatija, odnosno neuroloških sindroma. Kompletnu informaciju bi davao neonatalni skrining koji uključuje i ispitivalje funkcije slušnog nerva automatskim evociranim potencijalima moždanog stabla.
Negativan rezultat skrininga ne znači da dete obavezno ima oštećenje sluha. Štaviše, najčešće su razlozi veoma bezazleni poput postojanja sirastog maza u spoljašnjem slušnom hodniku ili embrionalnog tkiva u srednjem uvu. Upravo zato se ponavlja bar dva puta pre nego što se odojče uputi na kompletnu audiološku obradu.
Osnovni cilj što ranijeg otkrivanja senzorineuralnog oštećenja sluha kod dece jeste započinjanje pravovremene i odgovarajuće rehabilitacije kojom se smanjuju posledice oštećenja sluha, kao i stvaranje pretpostavki za eventualnu primenu drugih metoda zbrinjavanja ovog senzornog nedostatka.
Dijagnoza senzorineuralnog oštećenja sluha se postavlja na osnovu subjektivnih i objektivnih metoda za ispitivanje sluha. Procedura za postavljanje dijagnoze senzorineuralnog oštećenja sluha pored otorinolaringološkog pregleda obuhvata sledeće korake: posmatranje deteta i objektivna snimanja evociranih potencijala moždanog stabla i tzv. snimanje evociranih potencijala u mirnom stanju. Ista nisu neprijatna, nisu bolna, jedino traže da dete bude opušteno, mirno, najbolje u prirodnom snu ili ako nije moguće drugačije, u sedaciji.
Senzorineuralno oštećenje sluha se, u pravom smislu ne leči, ali postoje načini tehnološke i stručne podrške, koji omogućavaju da dete stekne sposobnost slušanja, razumevanja i razvoja govora, naravno, ukoliko nije udruženo sa drugim poteškoćama u psihofizičkom razvoju.
Dete dobija adekvatan slušni amplifikator (pojačivač zvuka) ili potom kohlearni implant operativnim putem, u zavisnosti od stepena oštećenja sluha. Sve to se obavlja uz pažljivu timsku pripremu. U našoj ustanovi se nalazi najveći Centar za selekciju kandidata za kohlearnu implantaciju u dečjoj populaciji od 2002. a poslednjih 13 godina se sprovodi i navedena operacija.
Redovne kontrole su na par meseci ili po potrebi.
Trajno senzorineuralno oštećenje sluha jeste doživotni nedostatak i ne može se nikada poboljšati, može samo uznapredovati ukoliko je progresivno po prirodi.
Ogroman broj senzorineuralnih oštećenja sluha nije moguće sprečiti jer su posledica faktora rizika koji se ne mogu sprečiti, kao što su npr. recesivno nasleđena oštećenja koja se javljaju po prvi put u porodici, prevremeno rođena deca lečena u odeljenjima inetnzivne nege i slično.
Infektivne bolesti, poput nekih bakterijskih meningitisa, se mogu sprečiti već dvadesetak godina. Imunizacija je najbrža, najefikasnija i najekonomičnija mera prevencije obolevanja i umiranja od teških zaraznih bolesti. U Kalendaru vakcinacije u Srbiji je obavezna vakcina protiv Haemophilus influenzae B od septembra 2006, a protiv Pneumoccocus pneumoniae od 2018. godine.
Ako je reč o izolovanom oštećenju sluha,, pravovremenoj dijagnostici i adekvatnoj intervenciji, dete može imati kvalitetan život i otvorene mogućnosti za osvajanje maksimalnog postignuća u školovanju po redovnom programu.
Nezaobilazan značaj i uticaj na proces i ishod slušnogovorne rehabilitacije ima mogućnost postojanja udruženih poremećaja zdravlja. Naime, postoji preko 400 poznatih sindroma koji uključuju oštećenje sluha, postoje infektivni sindromi, kao i metaboličke bolesti koje u 80 % slučajeva sadrže i oštećenje sluha.