Close
Скрининг подразумева систематску примену медицинских тестова у циљу раног откривања болести, у тренутку када симптоми болести нису присутни. Ради се код особа које нису тражиле лекарску помоћ, ради откривања појединаца с ризиком од одређеног поремећаја који је довољно велики да оправдава даље испитивање и превентивно деловање. Пошто се испитивање спроводи пре него што особа испољи симптоме, лечење може да започне пре него што се развије клиничка слика и пре него што настану компликације.
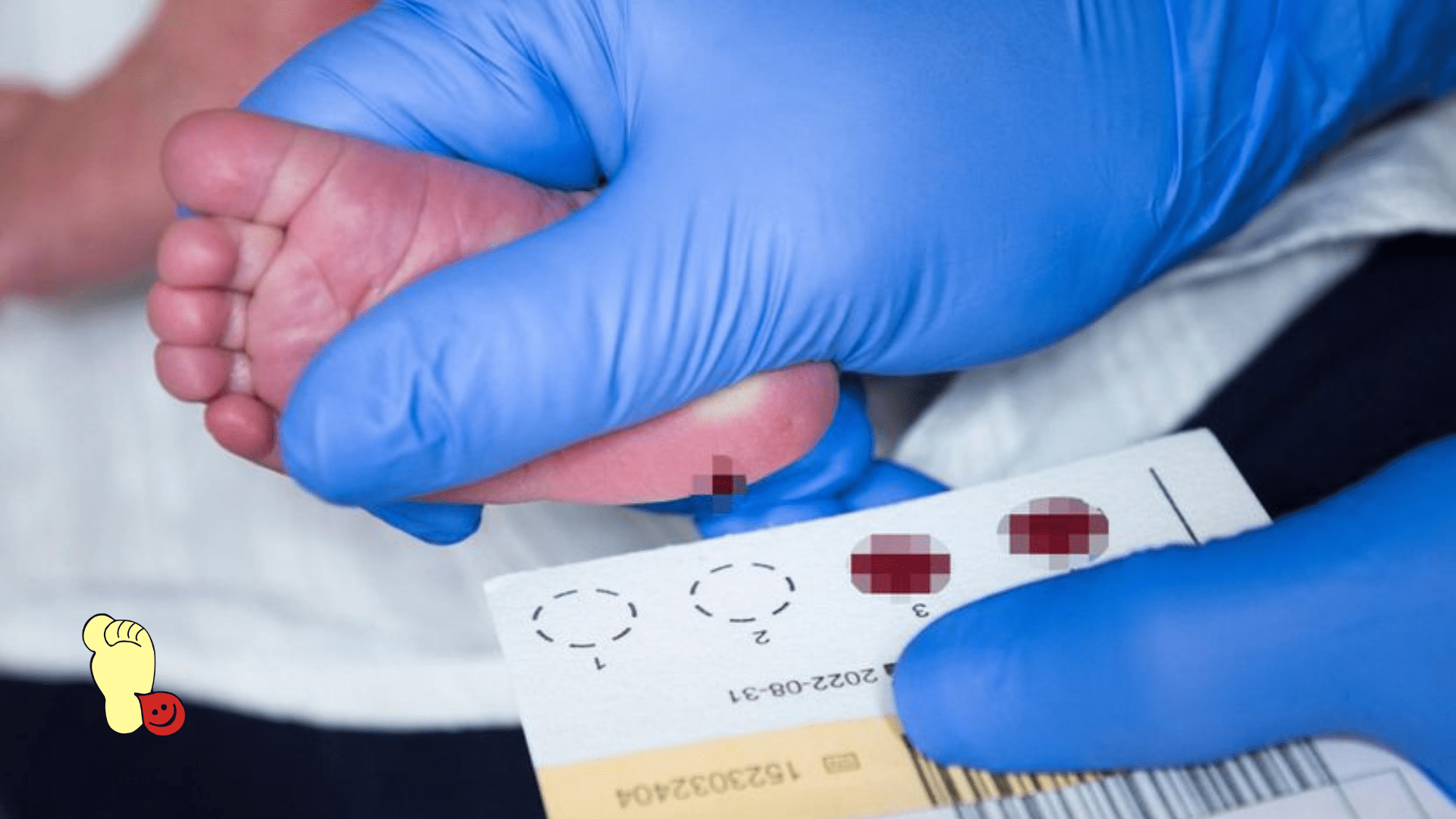
Сва деца рођена у Републици Србији пролазе неонатални скриниг. Крв за скрининг се узима у породилишту, а анализе се раде у лабораторијама ван породилишта. У Централној Србији узорци се шаљу у лабораторију Института за здравствену заштиту мајке и детета Србије „Др Вукан Чупић“, а у Војводини у лабораторију Института за здравствену заштиту деце и омладине Војводине. За децу из целе Србије, обрада узорака за скрининг на спиналну мишићну атрофију се врши на једном месту, тј. на Биолошком факултету у Београду.
Постоји велики број болести и стања за која се може спроводити неонатални скрининг. Број болести на које се спроводи скрининг разликује се од земље до земље.
Ако резултат неонаталног скрининга није нормалан, то још увек не значи да дете има болест за коју се скрининг врши. У том случају, неопходно је урадити додатне тестове, како би се утврдило да ли дете има болест за коју се скрининг спроводи. У већини случајева, деца која имају болест по рођењу изгледају потпуно нормално. Неонатални скрининг помаже да се болест открије правовремено, тако да терапија може да се започне што пре и на тај начин спрече компликације.
У Србији се спроводи неонатални скрининг анализом капи крви која се узима из пете новорођенчета у породилишту на 4 болести: фенилкетонурију, конгенитални хипотироидизам, цистичну фиброзу и спиналну мишићну атрофију. У току 2025. године, из исте капи, треба да започне скрининг и на конгениталну адреналну хиперплазију. Такође, у бројним, али не свим породилиштима, новорођеној деци се ради и скрининг слуха.
За ближе информације о обољењима, кликните на линкове испод.
Фенилкетонурија је болест у којој аминокиселина фенилалнин (саставни део свих протеина) не може да се разгради, што доводи до оштећења нервног система, озбиљне менталне заосталости и епилепсије.
Узрок је генетички. Здрави родитељи носе склоност за обољевање, а вероватноћа у свакој трудноћи да дете оболи је 25%. Ако се деси, генетички поремећај доводи до смањене активности ензима фенилаланин хидроксилазе који је неопходан за разлагање фенилаланина.
Новорођена деца са фенилкетонуријом немају симптоме болести и не разликују се од остале здраве новорођенчади. Ако болест не почне да се лечи у првом месецу живота, први симптоми се јављају тек после неколико месеци живота у смислу неуролошког пропадања.
Фенилкетонурија се открива непосредно по рођењу, у оквиру неонаталног скрининг програма, у Србији и готово у свим земљама света. Организовано откривање болести у нашој земљи траје више од четрдесет година. Узорак крви новорођеног детета се у трећем дану живота (најраније 48 сати од рођења, искључиво у породилишту), када смо сигурни да је дете јело јер је то једини начин да се фенилаланин унесе у организам. Узорци крви на филтер папиру се шаљу у централну лабораторију где се одређује концентрација фенилалнина. Резултати анализа се чувају у архиви централне лабораторије, као и у породилишту, на начин који гарантује поштовање принципа лекарске тајне.
У просеку се у Србији открије два до три детета годишње са фенилкетонуријом.
У случају да су резултати испитивања нормални, родитељи се о томе не обавештавају.
Ако резултат покаже више концентрације фенилалнина то још увек не означава болест, али се та деца позивају у Институт ради провере и допунских анализа.
Да, и то веома успешно. Наиме, симптоми фенилкетонурије се не јављају ако се терапија почне на време и добро спроводи.
Фенилкетонурија се лечи дијетом. Дијета подразумева ограничени унос протеина уз
коришћење мешавина аминокиселина без фенилалнина. Дијета мора бити контролисана и
вођена од стране стручних лица која се баве фенилкетонуријом. Родитељи морају да се добро
обуче у спровођењу дијете.
Контроле се углавном обављају слањем узорака крви на филтер папиру (мерењем
концентрације фенилалнина), а ређе доласком у Институт на амбулантни преглед.
Као свака друга генетичка болест фенилкетонурија траје цео живот, те се препоручује доживотна дијета која с временом постаје мање захтевна.
За сада се фенилкетонурија не открива пре рођења, с обзиром да након постављене ране
дијагнозе могу да се спрече симптоми посебно организованом исхраном.
Стриктно придржавање дијете омогућава нормалан раст и развој, а с временом дијета постаје
саставни део живота сваке особе са фенилкетонуријом. Фенилкетонурија није препрека
нормалном животу.
Конгенитални хипотироидизам је стање у коме у организму нема довољно хормона штитасте жлезде. Беба се рађа са овим стањем (реч „конгенитално“ значи урођено, тј. да је особа рођена са тим).
Штитаста (тироидна) жлезда налази се у пределу врата и она лучи тироидне хормоне. Ови хормони важни су за правилан раст и развој, одржавање телесне температуре, као и за бројне метаболичке процесе. Уколико штитаста жлезда не лучи довољно хормона говоримо о „хипотироидизму“.
Узроци могу бити разни – штитаста жлезда може да недостаје, може бити мања него уобичајено или формирана, али на другом месту од уобичајеног. Понекад, проблем може да буде проузрокован и променама на генетичком нивоу. Тада поремећај може да се види код више чланова једне породици.
Поред тога, смањена количина хормона штитасте жлезде може се наћи код новорођенчади чије мајке не користе довољно јода у исхрани (што је у Србији изузетно ретко) или код оних чије мајке узимају прекомерне количине јода или одређене лекове. У тим случајевима обично дође до постепеног опоравка.
Највећи број новорођенчади нема никакве симптоме и уобичајено се понашају. Већина новорођенчади у прве 2-3 недеље има довољно хормона штитасте жлезде који су кроз постељицу прешли из крви мајке у крв детета.
Ако симптоми постоје, то може бити нешто од следећег: жутица, мања активност (беба неуобичајено дуго спава), успорени или ретки покрети, промукао плач, проблеми са храњењем, констипација (затвор), неуобичајен изглед – увећан језик, напетост око пупка, широко отворена велика фонтанела, отворена мала фонтанела, смањен мишићни тонус (хипотонија), сува кожа, хладни периферни делови руку или ногу.
У Србији (као и већини земаља у свету) се од 1983. године спроводи неонатални скрининг на конгенитални хипотироидизам. Он подразумева узимање једне капи крви из пете новорођенчета и одређивање вредности хормона из тог узорка. Узорак крви се узима најраније у 48. сату живота, искључиво у породилишту. Резултати анализа се чувају у архиви централне лабораторије, као и у породилишту, на начин који гарантује поштовање принципа лекарске тајне.
Ако се рано (одмах по дијагностиковању) започне са давањем терапије, спречавају се могуће последице и дете се у потпуности нормално развија. Уз терапију, деца и одрасли са конгениталним хипотироидизмом имају сасвим нормалан живот.
Веома је важно да се одржавају нормални нивои хормона штитасте жлезде у свим узрастима, али је то нарочито важно код одојчади и мале деце зато што су хормони штитасте жлезде врло битни за раст и развој детета, као и за функционисање и развој мозга, те је неопходно да се родитељи стриктно придржавају терапије коју је прописао ендокринолог.
Конгенитална адренална хиперплазија (КАХ) је урођени поремећај надбубрежних жлезда који доводи до смањене продукције одређених хормона и прекомерне продукције других. Беба се са овим поремећајем рађа, а КАХ најчешће настаје због урођеног недостатка ензима који је неопходан за нормалну синтезу хормона у надбубрежним жлездама.
Надбубрежне жлезде се налазе изнад оба бубрега и луче неколико веома важних хормона као што су кортизол, алдостерон и андрогени (мушки полни хормони). Код КАХ-а, најчешће недостаје ензим 21-хидроксилаза, што доводи до недостатка кортизола и/или алдостерона и великог вишка мушких полних хормона.
Постоји више облика КАХ-а, али најтежи је такозвани „облик са губитком соли“. Симптоми се могу јавити у првим данима живота и укључују: повраћање, лоше напредовање, поспаност, одбијање оброка, дехидрацију, лоше опште стање.
Код девојчица је већ на рођењу уочљив другачији изглед спољашњих гениталија, које често личе на мушке због великог вишка мушких полних хормона. Код дечака спољашње гениталије су типичне мушке, па се болест открива касније, али углавном у прве 2 недеље живота.
Не може се спречити, јер је генетски поремећај, али се могу спречити компликације раним откривањем и правилним лечењем.
Цистична фиброза је наследна болест која захвата више органских система у телу, али највише плућа, панкреас и јетру. Болест настаје услед поремећаја рада јонског канала који је задужен за пролазак хлора кроз мембрану ћелија.
Највећи број новорођенчади нема посебне симптоме и делују потпуно здраво.
Понекад се дешава да густ цревни садржај блокира нормалан пролазак столице кроз црева изазивајући њихову опструкцију и надимање стомака, стање које се назива меконијални илеус. Оно захтева хируршко лечење већ у првим данима живота и јасно побуђује сумњу код лекара да новорођенче болује од цистичне фиброзе.
Бебе оболеле од цистичне фиброзе могу имати продужену неонаталну жутицу, чешће респираторне инфекције или слабије напредовање у телесној маси, уз појаву маснијих столица.
Промене на плућима, панкреасу и јетри су почеле да се развијају код бебе док је још била у мајчином стомаку, стога је од велике важности поставити дијагнозу цистичне фиброзе и пре него што се јаве специфични респираторни или цревни симптоми, да би се одговарајућа терапија започела што раније. На време примењена терапија успорава природни ток болести и спречава рани настанак промена на поменутим органима бебе.
У нашој установи се од априла 2022. године спроводи неонатални скрининг на цистичну фиброзу, из узорка капиларне крви на филтер папиру узетог из пете новорођенчета док је боравило у породилишту. Овај узорак се из свих породилишта у Србији шаље у Институт на даљу анализу. Изузетак су породилишта из аутономне покрајине Војводина, која самостално спроводи неонатални скрининг на својој територији.
У случају да је овај први налаз позитиван, о томе обавештавамо породилиште из ког је први узорак и послат. Након контактирања породице саветује им се долазак у Институт ради понављања анализе из новог узорка капиларне крви на филтер папиру, најкасније до 21-ог дана живота новорођенчета. Уколико и овај други налаз буде позитиван, то значи да постоји вероватноћа да беба заиста и болује од цистичне фиброзе.
За постављање дефинитивне дијагнозе потребан је допунски знојни тест, којим се анализира концентрација хлорида у зноју бебе. Тест се изводи касније, од трећег месеца живота, о чему родитељи буду на време обавештени путем телеграма. У њему се јасно наводи да је разлог њиховог позивања сумња да постоји вероватноћа да њихово дете болује од цистиче фиброзе, као и датум заказаног тестирања и име лекара који ће их тог дана примити.
Ако резултат знојног теста потврди сумњу да је беба оболела од цистичне фиброзе раде се и додатне молекуларне генетске анализе из узорка венске крви да би се утврдиле тачне мутације гена код бебе. Ова допунска генетска анализа је неопходна ради даљег планирања лечења ових пацијената.
Спинална мишићна атрофија (СМА) је урођено обољење мишића и нерава. Код овог обољења долази до раног пропадања нервних ћелија које инервишу мишиће, а зову се моторни неурони. Оне се налазе у кичменој мождини и у мозгу. Због ране смрти моторног неурона долази до пропадања мишића и њихове атрофије, односно губитка мишићне масе и снаге. Током времена болест напредује, због чега СМА спада у групу тешких, прогресивних неуромишићних болести. То је наследна болест и потребно је да разумемо њену генетичку основу, како би нам била јаснија природа обољења и различитост у испољавању.
Спинална мишићна атрофија је проузрокована недовољном функцијом протеина неопходног за преживљавање моторног неурона, чија је скраћеница СМН протеин од енглеског назива “survival motoneuron protein“. Овај протеин настаје преписивањем гена који има исту ознаку, СМН ген и налази се на 5. хромозому. Код здравих особа постоје два СМН гена, који се означавају као СМН1 и СМН2 ген. По правилу, постоје две копије СМН1 гена и више (2 до 6) копија СМН2 гена. Преписивањем СМН1 гена настаје 100% функционалан СМН протеин, значајан за живот моторних нервних ћелија. Преписивањем другог, тзв. СМН2 гена настаје протеин који је слабо функционалан. Само 10-15% овог протеина има активну функцију у животу нервних ћелија. Људи који болују од СМА имају „грешку“ у обе копије СМН1 гена, једне наслеђене од оца и друге, наслеђене од мајке. Та грешка је код 97-98% оболеле деце у виду недостатка неког сегмента СМН1 гена, стручно речено делеције у самом гену. Много ређе (код 2-3% оболелих) реч је о малој, тачкастој мутацији, која може „промаћи“ класичним генетичким тестовима и дијагностикује се тек са појавом симптома болести другачијим техникама (методом секвенцирања гена). С обзиром да СМН2 ген има „слабу“ функцију, он никако не може да замени СМН1 ген и потпуно надомести његову улогу, иако оболели могу имати већи број копија овог СМН2 гена. Међутим, значај СМН2 гена је у одређивању тежине болести, што зависи од броја његових копија.
Уколико особа има један нормалан СМН1 ген, а други са грешком, тј. недостатком једног дела, она неће имати болест. Такве људе називамо здравим преносиоцима болести. Ако оба родитеља носе ген са грешком, ризик да се роди дете оболело од СМА износи 25%. Могућност да добију дете које има само једну копију СМН1 гена и преносилац је болести као и сами родитељи износи 50%. Учесталост људи у општој популацији који су здрави преносиоци болести није мала и износи 1 на 40-60 особа.
СМА тип 0: Деца на рођењу углавном немају знаке болести. Ретко деца са СМА могу имати почетак болести пре рођења и да већ на рођењу испоље млитавост (хипотонију), мишићну слабост, ослабљене спонтане покрете екстремитета. Они врло брзо развијају поремећај дисајне функције, до немогућности самосталног дисања и живе кратко. За ову децу кажемо да имају „нулти“ тип СМА, који се издваја као пети облик ове болести и јавља се код око 1% оболелих. 2
СМА тип 1 (Вердниг-Хофманова болест): Највећи број оболелих, око 60%, има СМА тип 1 или Вердниг-Хофманову болест, која је добила име по ауторима који су је међу првима описали. Ова деца се рађају без симптома и знакова болести. Први знаци болести се јављају у првих 6 месеци живота, а најчешће у 2.-3. месецу. Слабост код нелечене деце врло брзо напредује, најпре се смањује спонтана покретљивост ногу, а потом руку и мишића трупа, важних за дисање, тако да деца не могу самостално да дишу и потребна им је потпора машином за дисање. Имају тешкоће са гутањем хране. Никада не стекну способност самосталног седења. Док није било лекова, деца са СМА тип 1 су умирала пре 2. рођендана. Они имају по 1 до 2 копије СМН2 гена.
СМА тип 2 или Дубовицева болест: Ако губитак стечених моторичких функција код детета услед слабости мишића наступи од 7. до 18. месеца живота, говоримо о типу 2 СМА. Ова деца науче да седе, али никада не проходају. Код најтежих форми СМА тип 2, деца током времена губе функцију седења. Посебан проблем за њих представљају деформитети кичменог стуба. Њихов животни век је дужи од 2 године, а око 70% живи до 25. године живота. Они углавном имају по 3 копије СМН2 гена.
СМА тип 3 или Кугелберг–Веландерова болест: Деца која имају 3–4 копије СМН2 гена проходају, али ход постаје нестабилан услед развоја мишићних слабости у периоду после 18. месеца живота. Ако се симптоми испоље од 18. до 36. месеца, говоримо о типу 3А болести, а уколико настану после 36. месеца живота, у питању је 3Б подтип болести. Болесници са СМА тип 3 имају нормалан животни век.
СМА тип 4 или адултни облик СМА: У питању су одрасли пацијенти, код којих тегобе у виду слабости доњих екстремитета, тешкоћа са стајањем и ходом настају од 20. до 30. године. Функцију самосталног хода углавном сачувају. Просечни животни век ових болесника се не разликује од опште популације и они поседују по 4 или више копија СМН2 гена.
У Србији, као и у већини земаља у свету, до 2023. године болест се дијагностиковала на основу клиничког прегледа детета. Код сумње на СМА деца су тестирана генетичким тестовима, ради потврде клиничке дијагнозе. Ови тестови су се радили у две референтне лабораторије у Србији, у генетичкој лабораторији Института за здравствену заштиту мајке и детета Србије и на Биолошком факултету Универзитета у Београду, као и данас. Од 15. септембра 2023. године спроводи се скрининг новорођенчади у свим породилиштима у Србији на СМА, узимањем узорка крви на филтер папиру који се шаље у Биолошки факултет Универзитета у Београду. У генетичкој лабораторији Биолошког факултета се ради генетичко тестирање и ово је једина болест код нас где је неонатални скрининг заправо генетички тест. Резултати тестирања се чувају у архиви централне лабораторије, као и у породилишту, на начин који гарантује поштовање принципа лекарске тајне.
Да, могуће је добити лажно негативан налаз. То се дешава у ситуацијама када особа поседује само једну уместо две копије СМН1 гена, и у тој једној копији поседује тачкасту мутацију, која „руши“ нормалну функцију овог гена, односно протеина добијеног његовим преписивањем. Та могућност износи 2–3%. У овим ситуацијама болест се открива са појавом првих симптома.
У случају да су резултати тестирања негативни, односно нормални, родитељи се не обавештавају о томе.
Ако резултат указује да постоји СМА код бебе, анализа се још једном понавља, како би се са сигурношћу потврдило обољење. Одређује се и број копија СМН2 гена, што је значајно за тежину обољења, односно указује на то који ће се тип болести развити код детета. Родитељи се позивају на разговор и дијагнозу им саопштава лекар, неуропедијатар или дечији неуролог који се бави лечењем болесника са СМА.
Спинална мишићна атрофија се јавља код једног од 6 000 до 10 000 живорођене деце. Захваљујући неонаталном скринингу, у Србији је процењено да једно од 9066 новорођене деце болује од СМА. Дакле, у просеку се на годишњем нивоу роди 5–6 беба са овом болешћу.
Постоје лекови за лечење СМА, који још увек нису терапија потпуног излечења, већ мењају
природни ток болести. Захваљујући новим, иновативним лековима и најтежи болесници са
СМА тип 1 достижу поједине фазе моторичког развоја, као што су седење, стајање и ход, што је
ранијих деценија било незамисливо. Започињањем терапије веома рано, пре првих симптома
болести, даје се оболелом детету могућност бољег живота, са мање потешкоћа и
компликација. Сваки дан кашњења у терапији значи умирање нових моторних неурона и
развој слабости мишића.
За децу са СМА је најважније да се лечење започне што раније, пре него ли се појаве први знаци мишићне слабости. Често чујемо крилатицу, која је тачна, да је „моторни неурон = време“, која одражава важност раног започињања терапије.
Први лек који је одобрен за терапију СМА је нусинерсен. Његова примена у Америци је започета 2016. године, у Европи 2017. године, а у Србији јула 2018. године. То је мали молекул (тзв. олигонуклеотид), који се апликује у кичмени канал и директно делује на моторне неуроне предњих рогова кичмене мождине, спречавајући њихово изумирање. Он побољшава функцију СМН2 гена, делујући да се синтетише много више функционалног СМН2 протеина, у циљу да се надомести непостојећи СМН1 протеин.
Други лек слично делује као и нусинерсен, зове се рисдиплам и примењује се у виду сирупа.
Трећа доступна терапија је генска терапија („Золгенсма“), која се примењује у виду једнократне инфузије.
Не смемо заборавити значај физикалне терапије и мултидисциплинарни приступ у лечењу деце са СМА, без чега примена медикаментне терапије неће дати задовољавајуће резултате.
У најранијем узрасту неуролошке контроле су честе, а зависе од клиничког облика болести и општег стања детета. Веома је важно да се прате фазе моторичког развоја детета. Оболели од СМА имају нормалну интелигенцију, могу бити и натпросечно интелигентни.
Спинална мишићна атрофија је генетичко обољење, записано у генетском коду особе која га носи и самим тим је болест доживотна.
Спинална мишићна атрофија може да се превенира у породици која има оболело дете, тако што ће се урадити пренатално генетичко тестирање у првим месецима трудноће мајке. Трудница треба да се на време обрати генетичком саветовалишту.
Дете са СМА може имати далеко бољу прогнозу за развој и живот, захваљујући примени нових, иновативних лекова, који модификују природни ток болести и преводе тежи у лакши облик болести.
Појам оштећења слуха обухвата наглувост и глувоћу. Наглувост подразумева ограничење слушне способности, а глувоћа одсуство слушне функције. Оштећења слуха обухватају широк спектар, али најважнија су трајна, конгенитална, тј. урођена или рано настала, обострана тешког степена. Трајно оштећење слуха код деце подразумева обострано сензоринеурално оштећење слуха са прагом слуха испод 40 дБ (средња вредност за чисти тон на карактеристичним фреквенцијама). Оштећења слуха могу бити једнострана и обострана, могу бити пролазна и трајна, наследна и стечена а у односу на природу прогресивна и непрогресивна. Зависно од времена настанка односно испољавања могу бити пренатална, перинатална и постнатална. У зависности од времена испољавања у односу на развој говорне функције, оштећења слуха деле се на прелингвална, настала пре развоја говора и постлингвална, настала полсле успостављања говорне функције.
Сензоринеурално оштећење слуха (СНОС) је последица оштећења финог сензорног ткива унутрашњег ува или читавог слушног нерва. Бројни су фактори ризика за настанак оштећења слуха. Прихваћена је следећа класификација:
Оштећење слуха се у већини случајева не види. Новорођенче не показује никакве клиничке знакове, има уобичајено понашање. Тек се временом може приметити слабија реакција на звук.
Код малог броја деце се може рећи да је видљиво, заправо онда када одсуство или значајно умањење ушне шкољке указују на постојање оштећења слуха.
Урођено оштећење слуха се открива неонаталним скринингом. Скрининг се спроводи у породилиштима снимањем транзиторних отоакустичких емисија (ТЕОАЕ), које указују на постојање оштећења слуха у спољашњем, средњем и унутрашњем уву. Оно што оне не откривају, јесте оштећење слушног живца и и других слушних путева који воде информацију о звуку до коре великог мозга. Ови, последњи наведени поремећаји се јављају изузетно ретко. Мерење ОАЕ је објективно, неинвазивно, брзо, поновљиво и прецизно. У основи ОАЕ дају објективан увид у функционисање целог пресинаптичког аудитивног система. На срећу, највећи број слушних оштећења последица је патолошких промена управо у овом делу аудиторног апарата. Транзиторне ОАЕ, са широкопојасно фреквентним, краткотрајним звучним стимулусом који активира целу кохлеу јер у себи садржи све фреквенције, у најширој су употреби. Веома су осетљива метода и не могу се регистровати са оних подручја кохлее где је праг слуха већи од око 30 дБ. Отоакустичке емисије се у клиничкој пракси употребљавају више од 30 година. Овом методом се најбоље може испитати функција сензорних слушних ћелија и диференцирати њихово оштећење од оштећења проксималних нервних аудиторних структура и функција.
За новорођенчад и малу одојчад, до шест месеци, физиолошка мерења су приступ избора. Чак и чујуће, здраве бебе не обезбеђују реалне бихејвиоралне одговоре на звук пре навршених шест месеци старости. Старија деца, међутим, могу бити тестирана ефикасно и ефективно и физиолошким и бихејвиоралним мерењима.
Слушно оштећење не подлеже обавези пријављивања у Србији и то доводи до недостатка података о правом броју , али процена је да се годишње у Србији рађа око 150 деце са трајним оштећењем слуха. Пре свега се мисли на трајно сензоринеурално оштећење слуха (TSNOS). Његово откривање и јесте циљ неонаталног скрининга. Оштећење слуха представља најчешће сензорно оштећење у људи, са значајним социјалним и психолошким импликацијама. Процењено је да приближно 20% људске популације старије од 18 година пати од неке форме слушног оштећења.
На нивоу опште популације, троје од 1000 новорођене деце има оштећење слуха, а од њих једно има дубоко конгенитално трајно сензоринеурално оштећење слуха, а у популацији деце која су боравила у одељењима за интензивну негу та учесталост износи 1:50.
Нормалан резултат скрининга који се спроводи само снимањем отоакустичких емисија указује на то да је уредна функција спољашњег, средњег и унутрашњег ува. О функцији слушног нерва не даје информацију. То значи да потврда уредне функције слуха није комплетна. Не открива се аудитивна неуропатија, спектар поремећаја, посебан сензоринеурални ентитет, који се јавља знатно чешће него што се раније сматрало, чак у око 10 % пацијената са трајним сензоринеуралним оштећењем слуха. Хипербилирубинемија (жутица) и хипоксија (мањак кисеоника) у новорођеначком узрасту су најважнији ризико фактори за њен настанак. Код неке деце је то наследни поремећај, а јавља се у склопу полинеуропатија, односно неуролошких синдрома. Комплетну информацију би давао неонатални скрининг који укључује и испитиваље функције слушног нерва аутоматским евоцираним потенцијалима можданог стабла.
Негативан резултат скрининга не значи да дете обавезно има оштећење слуха. Штавише, најчешће су разлози веома безазлени попут постојања сирастог маза у спољашњем слушном ходнику или ембрионалног ткива у средњем уву. Управо зато се понавља бар два пута пре него што се одојче упути на комплетну аудиолошку обраду.
Основни циљ што ранијег откривања сензоринеуралног оштећења слуха код деце јесте започињање правовремене и одговарајуће рехабилитације којом се смањују последице оштећења слуха, као и стварање претпоставки за евентуалну примену других метода збрињавања овог сензорног недостатка.
Дијагноза сензоринеуралног оштећења слуха се поставља на основу субјективних и објективних метода за испитивање слуха. Процедура за постављање дијагнозе сензоринеуралног оштећења слуха поред оториноларинголошког прегледа обухвата следеће кораке: посматрање детета и објективна снимања евоцираних потенцијала можданог стабла и тзв. снимање евоцираних потенцијала у мирном стању. Иста нису непријатна, нису болна, једино траже да дете буде опуштено, мирно, најбоље у природном сну или ако није могуће другачије, у седацији.
Сензоринеурално оштећење слуха се, у правом смислу не лечи, али постоје начини технолошке и стручне подршке, који омогућавају да дете стекне способност слушања, разумевања и развоја говора, наравно, уколико није удружено са другим потешкоћама у психофизичком развоју.
Дете добија адекватан слушни амплификатор (појачивач звука) или потом кохлеарни имплант оперативним путем, у зависности од степена оштећења слуха. Све то се обавља уз пажљиву тимску припрему. У нашој установи се налази највећи Центар за селекцију кандидата за кохлеарну имплантацију у дечјој популацији од 2002. а последњих 13 година се спроводи и наведена операција.
Редовне контроле су на пар месеци или по потреби.
Трајно сензоринеурално оштећење слуха јесте доживотни недостатак и не може се никада побољшати, може само узнапредовати уколико је прогресивно по природи.
Огроман број сензоринеуралних оштећења слуха није могуће спречити јер су последица фактора ризика који се не могу спречити, као што су нпр. рецесивно наслеђена оштећења која се јављају по први пут у породици, превремено рођена деца лечена у одељењима инeтнзивне неге и слично.
Инфективне болести, попут неких бактеријских менингитиса, се могу спречити већ двадесетак година. Имунизација је најбржа, најефикаснија и најекономичнија мера превенције оболевања и умирања од тешких заразних болести. У Календару вакцинације у Србији је обавезна вакцина против Haemophilus influenzae B од септембра 2006, а против Pneumoccocus pneumoniae od 2018. године.
Ако је реч о изолованом оштећењу слуха,, правовременој дијагностици и адекватној интервенцији, дете може имати квалитетан живот и отворене могућности за освајање максималног постигнућа у школовању по редовном програму.
Незаобилазан значај и утицај на процес и исход слушноговорне рехабилитације има могућност постојања удружених поремећаја здравља. Наиме, постоји преко 400 познатих синдрома који укључују оштећење слуха, постоје инфективни синдроми, као и метаболичке болести које у 80 % случајева садрже и оштећење слуха.
Институт за здравствену заштиту мајке и детета Србије ,,Др Вукан Чупић’’
Copyright © 2025. Сва права задржана.